
全程导医网 徐州就医信息:2024年2月29日是第十七届国际罕见病日。今年的主题是“关注罕见、点亮生命之光,弱有所扶、践行人民至上”。
今天让我们一起关注关爱罕见病群体,让“罕见”被看见。
2023年10月,一对眼泪汪汪的父母推着轮椅来到徐州医科大学附属医院神经内科葛巍医生的诊室,轮椅上坐着一个15岁的小姑娘一直在傻傻地笑。葛巍医师仔细询问病史方知:这位小姑娘叫燕子(化名),打小是成绩优异、兴趣广泛的“学霸”。然而到了9岁左右时,奇怪的事情发生了,燕子的成长就像被按了“倒退键”:注意力不集中、走路容易跌跤、智力逐渐退化,还时伴有肢体抽搐、间断痴笑、语言能力渐渐丧失。
这究竟是怎么了?!

葛巍医生经查体发现,燕子除了智力退化及语言功能丧失外,还存在高弓足,马蹄内翻足畸形(图1),双下肢肌张力高,四肢腱反射亢进等。详细询问孩子父母得知,燕子11岁的弟弟智力水平也不如同龄孩子。由此不排除遗传性疾病的可能。考虑患者家庭经济情况,在完善相关检查后科室为其申请了中国罕见病联盟的公益金支持的免费基因检测项目。
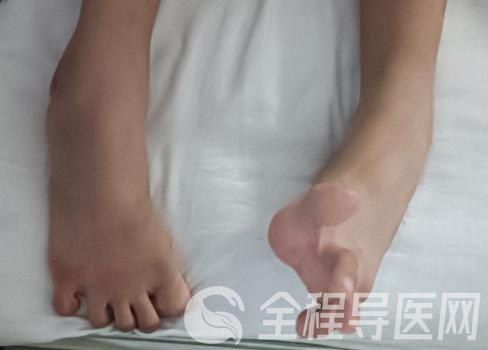
图1 患者表现为马蹄内翻足畸形
测序结果回复:孩子NPC1基因存在两处突变,分别为c.2292G>A同义突变和c.3591+2dup重复突变(见图2),后经Sanger验证两处变异分别来自父母,其弟弟携带相同变异,根据ACMG遗传变异分类标准与指南,两处变异均评级为可能致病的。家系图如图3所示。真相终于大白,按下孩子成长“倒退键”的原来是尼曼匹克病(C型)。
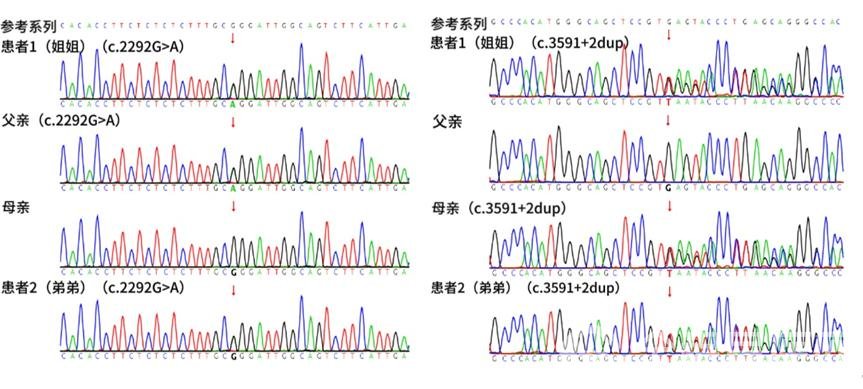
图2 NPC1基因复合杂合突变导致NPD-C患者家系的全基因组高通量测序验证图(红色箭头提示突变位点所在位置),c.2292G>A源自父亲,c.3591+2dup源自母亲。
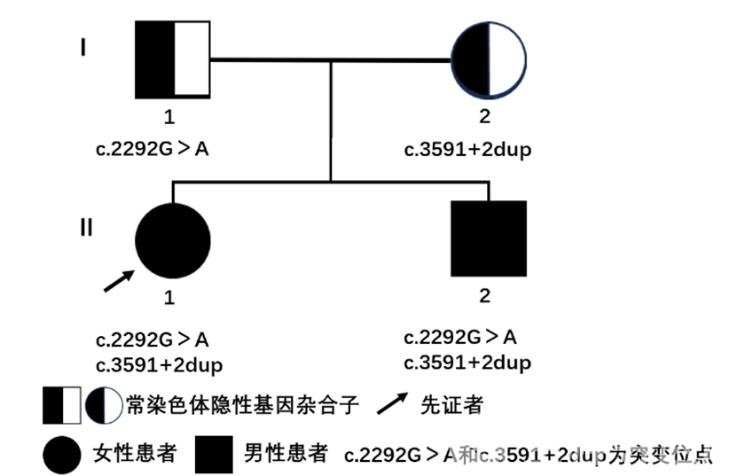
图3 NPC1基因杂合突变导致的常染色体隐性遗传的NPD-C疾病家系图
什么是尼曼匹克病?
尼曼匹克病(Niemann Pick disease,NPD)为常染色体隐性遗传疾病,主要分为A、B、C三种亚型[1],其中A、B亚型是由编码酸性鞘磷脂酶的SMPD1基因发生突变所致,酸性酯酶的作用为分解细胞膜上的鞘磷脂,缺乏该酶会导致鞘磷脂在细胞内积聚,又称酸性鞘磷脂酶缺乏症[2];C型(NPD-C)是由于NPC1(约95%)和/或NPC2(约5%)基因突变所致,使得结合和运输胆固醇的蛋白质功能缺陷,细胞内胆固醇堆积,继而影响细胞正常功能,尤其神经系统最易受累[1]。
NPD-C的常见临床表现包括认知障碍、共济失调、垂直注视麻痹、肌张力障碍、肝脾肿大、吞咽困难、肺功能受损等[1]。婴幼儿期最早的症状之一是肝脾肿大;儿童期和青少年期多表现为言语和运动能力落后;青少年期及成人期神经系统症状进一步加重,如认知障碍、共济失调、肌张力异常。
虽然目前针对NPD-C病因缺乏有效的治疗方法,但葡萄糖神经酰胺合成酶抑制剂麦格司他,是中国唯一获批准治疗NPD-C的药物且2020年被纳入国家医保,可稳定、改善患者进行性加重的神经症状,延缓病情的进展,被国内外多个权威指南推荐。

确诊后,医疗团队立即给予燕子和她弟弟进行药物干预治疗。2024年1月(治疗1月后)电话随访,燕子已经会喊“爸爸、妈妈”了;2024年2月(治疗2月后)随访获悉,燕子已经会哼唱歌曲“小苹果”了!“每一次听到孩子喊爸爸、妈妈,每一次看到孩子的病情改善,我们都充满了希望”,电话的那头孩子妈妈难以抑制内心的激动!
NPD-C的确诊需要临床医师进行详细的病史询问、细致的体格检查,一旦临床怀疑NPD,即便没有明确的家族史,也可考虑进行基因的筛查。早发现,早确诊,早干预,规范化的诊疗对NPD-C患者的预后有很大影响。
参考文献
[1] 国家卫生健康委罕见病诊疗与保障专家组. 罕见病诊疗指南(2019版),2019.
[2] Angeli O, Nagy Z, Schneider M. Ocular manifestation of an adult Niemann-Pick disease type B. Orvisi Hetilap[J] 2023; 164 (46): 1838-1844.
[3] Vanier, MT. Niemann-Pick diseases. Handb Clin Neurol. 2013; 113 1717-21. doi: 10.1016/B978-0-444-59565-2.00041-1
[4] 张安安, 刘晓琴, 吴涛. 尼曼匹克病C型诊治进展. 国际遗传学杂志[J] 2023; 46 (3): 216-222.
神经遗传及罕见病门诊
- 时间 -
每周日上午
- 地点 -
本部门诊四楼神经内科诊室
团队成员

葛 巍
主任医师
医学博士,副教授

许 刚
副主任医师
医学硕士

鲁淑谨
主治医师
医学博士(在读)

牛 牧
主治医师
医学博士(在读)

房效莉
主治医师
医学博士(在读)

贾 潇
主治医师
医学硕士
徐州导医热线:0516-85707122






